
|
|
|
|
|
GENETIK |
Anwendung der Gentechnik in der Kriminalistik (4) |
|

|
|
|
Identifizierung und Kartierung des GenomsGenotypisierung:Unter dem Begriff Genotypisierung versteht man die Anfertigung eines genetischen Profiles, eines genetischen Fingerabdruckes, der eine eindeutige Zuordnung zulässt, wie etwa: Aus welcher Zucht stammt dieses Tier, von welchem Saatgut stammt diese Pflanze oder von welchem Menschen stammt ein Bluttropfen. Diese molekulargenetische Identifikation kann nicht nur für phylogenetische Fragestellungen verwendet werden, diese Stammbaumanalysen sind beispielsweise für Zoos von Interesse. Es können Inzuchteffekte vermieden werden, und für die Wissenschaft wichtige Fragestellungen erfahren dadurch eine Klärung. Der Genetische Fingerabdruck erlaubt auch den Herkunftsnachweis. Eine gerade durch mehrere Skandale sensibilisierte Bevölkerung wird verstärkt wissen wollen, woher die Nahrungsmittel kommen und DNA-Analysen sind das Mittel der Wahl, um rasch und effizient diese dringenden Fragen zu beantwor-ten. Minisatelliten-Analyse (Southern Blot):Das wichtigste Werkzeug bei der Southern Blot-Analyse von RFLP-Systemen sind bestimmte Eiweißstoffe, Restriktionsenzyme genannt, die aus Bakterien isoliert werden, und die wie chemische Scheren die DNA an ganz bestimmten Stellen schneiden können. "RFLP" bedeutet "restriction fragment length polymorphism". Gemeint ist, dass jeder Mensch auf der Erde ein ganz spezielles Muster an DNA-Fragmentlängen aufweist. Als Erkennungsmuster für diese Enzyme dienen bestimmte Basensequenzen, die mehr oder weniger zufällig verstreut auf dem DNA-Molekül vorkommen. Wenn nun die DNA in den flankierenden Bereichen von Minisatellitensequenzen geschnitten wird, so entstehen DNA-Fragmente, deren Länge abhängig ist von der Anzahl der Minisatelliten-Wiederholungseinheiten. Bei der Southern-Blot-Analyse werden DNA-Fragmente, die durch Behandlung mit den schneidenden Enzymen entstanden sind, in einer Elektrophorese nach ihrer Länge getrennt, auf eine Membran übertragen und mittels radioaktiv oder Enzym-markierter "DNA-Sonden" die nur die gesuchten Fragmente erkennen können, sichtbar gemacht. So entstehen RFLP-Muster, deren Zusammensetzung typisch für ein einzelnes Individuum ist. Die beiden Merkmale eines jeden Genortes lassen sich dabei jeweils von der Mutter bzw. vom Vater ableiten. PCRDer Nachteil der DNA-Analysetechnik ist die Menge benötigter DNA (5-10 µg). Das ist bei Vaterschaftsfällen kein Problem. Bei winzigen Blut-, Sperma oder Speichelspuren liegt jedoch eine nicht überwindbare Mengenbeschränkung vor. Daher nutzen die Labors die Methode der DNA-Vervielfältigung durch die Po-lymerasekettenreaktion (polymerase chain reaction, PCR), für deren Entwicklung KARY MULLIS 1993 den Nobelpreis erhielt. Durch die Wahl geeigne-ter DNA-Startermoleküle wird bei der PCR-Typisierung nicht das gesamte Genom zerschnitten und aufgetrennt, sondern es werden ausgewählte DNA-Strecken vervielfältigt. Analyse mittels PCRPCR ist ein in vitro (Reagenzglas-) Verfahren zur gezielten Vermehrung von DNA mit Hilfe von synthetischen Oligonukleotiden und dem Enzym DNA-Polymerase. Durch wiederholte Zyklen wird aus einer DNA-Lösung ein spezifi-scher DNA-Abschnitt vermehrt. Synthetische Oligonukleotide (eine nur kurze Reihe von Basen auf der DNA) definieren den DNA-Abschnitt, der vervielfältigt werden soll. Sie werden PCR-Primer genannt, da nach ihrer Anlagerung an einen DNA-Einzelstrang von ihrem Ende ausgehend das Enzym DNA-Polymerase den komplementären Strang neu synthetisieren kann. Die Primer für ein bestimmtes PCR-System werden so gewählt, dass sie den zu untersuchenden Abschnitt der DNA einschließen. Damit die DNA-Neusynthese stattfinden kann, müssen neben den bereits erwähnten Primern und der Polymerase auch die vier verschiedenen reaktiven DNA-Bausteine, die so genannten Nukleotide, im Reaktionsgemisch vorhanden sein. Die eigentliche PCR-Reaktion findet in kleinen Reaktionsgefäßen
in einem so genannten Thermocycler statt. Als nächster Schritt wird das Reaktionsgemisch auf eine für die beiden Primer spezifische Temperatur zwischen 50°C und 65°C abgekühlt. Bei dieser Temperatur könnten sich die PCR-Primer an die DNA-Einzelstränge anlagern. Die Temperatur muss so gewählt werden, dass sich die Primer nur dort anla-gern, wo ihre Sequenz vollständig mit der des DNA-Einzelstrangs übereinstimmt. Im dritten Schritt des PCR-Zyklus, bei ca. 72°C, findet schließlich ausgehend von den Enden der Primer eine komplementäre Ergänzung der DNA-Einzelstränge statt, wodurch in diesem Bereich ein DNA-Doppelstrang entsteht. In jedem Folgezyklus findet nunmehr erneut eine Verdopplung dieses DNA-Abschnitts statt, wodurch es zu einer exponentiellen Vermehrung der Zielsequenz kommt. Typischerweise werden bei PCR-Vermehrung 28 - 32 Zyklen durchgeführt. Daraus errechnet sich theoretisch ausgehend von einer einzel-nen Kopie zu Beginn der PCR, dass am Ende nach 32 Zyklen 2 Milliarden Kopien des zu untersuchenden DNA-Abschnittes vorliegen. Tatsächlich kann man jedoch nicht mit einer 100%igen Verdopplung pro Zyklus rechnen. Realistische Werte liegen zwischen 70 und 90 %. Bei einer durchschnittlichen Effizienz von 80% errechnen sich nach 32 Zyklen 5 Mio. Kopien der Ausgangskopie der DNA. Dies reicht für eine Analyse mittels Elektrophorese. Kapillar-Elektrophorese:In der Gel - Elektrophorese wandern geladene, biologische Moleküle im elektrischen Feld. Die in Protonen und konjugierte Basen zerfallene DNA wandert vom negativen zum positiven Pol. Je nach Form und Größe wandern die die kleineren Moleküle, die die Gelporen schneller passieren, schneller als Größere. Die Analyse der PCR-Produkte, welche sich durch die Anwesenheit von Mikrosatelliten-Polymorphismen in ihrer Länge unterscheiden, erfolgt durch eine automatische Fragmentanalyse mittels Kapillar-Elektrophorese. Die DNA-Fragmente mit den Repeat-Merkmalen werden an der Kathode (-) auf-getragen und wandern bei Anlegen einer Hochspannung durch ihre eigene negative Ladung in die Kapillare in Richtung Anode (+) ein. In der Kapillare befindet sich ein flüssiges Polymer, dass wie ein Sieb die Fragmente sortiert und bei fortschreitender Wanderung durch die Kapillare auftrennt. Die PCR-Produkte (DNA-Teile) werden durch die Verwendung jeweils eines
PCR-Primers für jedes System sichtbar gemacht, der mit einem Fluoreszenzfarbstoff
(blau, grün oder gelb) markiert worden ist. Weiterhin wird ein mit
rotem Farbstoff markierter interner DNA-Längenstandard zugefügt,
der zur Messung der PCR-Produkte unbekannter Fragmentlänge benötigt
wird. Wandern nun die markierten DNA-Fragmente der zu untersuchenden Probe
aufgrund der Polarität des elektrischen Feldes der Elektrophorese
durch das in der Kapillare befindliche gelartige Polymer von der Kathode
zur Anode, werden sie der Länge nach aufgetrennt, d.h. die kleinen
Fragmente können schneller durch das Gel dringen als die großen
Fragmente. Sobald diese Fragmente am Fenster des Detektors vorbeiwandern,
werden die Farbstoffmoleküle durch einen Laser-strahl zur Fluoreszenz
angeregt. Diese Fluoreszenz wird nun durch ein optisches System auf einen
Detektor mit CCD-Kamera gebracht und dort nach Wellenlänge und Signalstärke
in digitale Messwerte umgewandelt.
Diese Werte werden schließlich mittels EDV-Analyse mit den bekannten Fragmentlängen des internen Standards verglichen und dann den Allelen der einzelnen DNA-Systeme gestaffelt nach Farbmarkierung und Fragmentlänge zugeordnet.
Abb.: Beispiel für DNA-Fragmente, die durch Elektrophorese getrennt wurden.
(Aus: http://www.uni-koblenz.de/~odsgroe/dnaanaly.php) |
|
|
Vorarlberger Bildungsserver |
| Anatomie/Physiologie | Botanik | Cytologie | Evolution | |
| Genetik | Humanbiologie | Ökologie | Sexualbiologie | Zoologie |
| Geschichte | Texte, Referate | Sehenswert | Kontakt | Physik |
Zwei Genetikthriller vom Autor dieser Seite |
.png)
Rudolf Oeller:Typhons RacheThriller über die vernichtende Kraft der Rache und den Traum vom ewigen Leben im Diesseits.
|
.png)
Rudolf Oeller:Typhon DistrictThriller über eine Gruppe junger Wissenschaftler, die an ihrer Gier zugrunde ging.
|

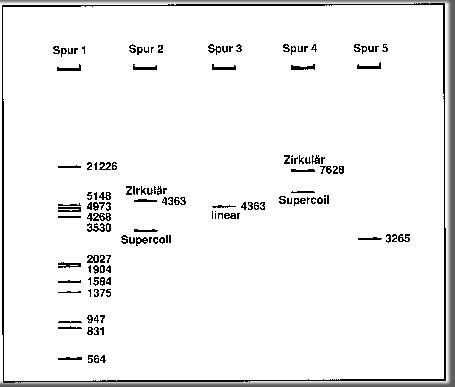 Spur
1 zeigt die Banden der DNA-Fragmente des DNA-Markers III, der durch
Schneiden des DNA-Fadens des Bakteriophagen Lambda (Lambda-DNA) mit dem
Restriktionsen-zymen Eco RI und Hind III entstanden ist. Den Banden sind
jeweils die Basenpaare (bp) der Fragmente zugeordnet.
Spur
1 zeigt die Banden der DNA-Fragmente des DNA-Markers III, der durch
Schneiden des DNA-Fadens des Bakteriophagen Lambda (Lambda-DNA) mit dem
Restriktionsen-zymen Eco RI und Hind III entstanden ist. Den Banden sind
jeweils die Basenpaare (bp) der Fragmente zugeordnet.